Identifying Unique Regions in Viruses using NCBI
My goal is to use some common tools and do some bioinformatics to identify interesting regions in Adenovirus genomes. Let's start with a high level plan:
- Get a bunch of adenovirus genomes, ideally ones that are as complete as possible.
- Do a MSA (multiple sequence alignment)
- From the MSA results, try to find regions with low mutations (conserved universal markers) or regions unique to a specific type of adenovirus (say, Adenovirus F).
- Scrutinize said regions - anything interesting?
Getting some Adenovirus
We can use the datasets command line tool from the NCBI. You can get datasets here.
It has a bunch of useful features -- including one specifically for finding viral genomes!
$ datasets summary virus --help
Print a data report containing virus genome metadata by accession or taxon. The data report is returned in JSON format.
Usage
datasets summary virus [flags]
datasets summary virus [command]
Available Commands
genome Print a data report containing virus genome metadata by accession or taxon
Global Flags
--api-key string Specify an NCBI API key
--debug Emit debugging info
--help Print detailed help about a datasets command
--version Print version of datasets
Use datasets summary virus <command> --help for detailed help about a command.
We want to search for genomes. That help screen is useful, too:
datasets summary virus genome --help
Print a data report containing virus genome metadata by nucleotide accession or taxon. The data report is returned in JSON format.
Refer to NCBI's [download and install](https://www.ncbi.nlm.nih.gov/datasets/docs/v2/download-and-install/) documentation for information about getting started with the command-line tools.
Usage
datasets summary virus genome [flags]
datasets summary virus genome [command]
Sample Commands
datasets summary virus genome accession NC_045512.2
datasets summary virus genome taxon sars-cov-2 --host dog
Available Commands
taxon Print a data report containing virus genome metadata by taxon (NCBI Taxonomy ID, scientific or common name for any virus at any tax rank)
accession Print a data report containing virus genome metadata by accession
taxon is the most useful command here, since we aren't look for a particular accession - just a general "give me the viruses".
We can do this as such:
datasets summary virus genome taxon "adenovirus" | json_pp > adenovirus.json
The resulting file has tons of information. I got about 900 entries, here's the general structure:
{
"accession" : "V01485.1",
"completeness" : "PARTIAL",
"length" : 179,
"nucleotide" : {
"sequence_hash" : "FF2602D4"
},
"release_date" : "1983-12-06T00:00:00Z",
"source_database" : "GenBank",
"submitter" : {
"names" : [
"Brinckmann,U.",
"Darai,G.",
"Flugel,R.M."
]
},
"update_date" : "1991-07-08T00:00:00Z",
"virus" : {
"lineage" : [
{
"name" : "Viruses",
"tax_id" : 10239
},
{
"name" : "Varidnaviria",
"tax_id" : 2732004
},
{
"name" : "Bamfordvirae",
"tax_id" : 2732005
},
// ...
{
"name" : "unclassified Mastadenovirus",
"tax_id" : 346173
},
{
"name" : "unidentified adenovirus",
"tax_id" : 10535
}
],
"organism_name" : "unidentified adenovirus",
"tax_id" : 10535
}
}
This sample only has a length of 179 - basically a fragment. An entire Adenovirus genome is in the ballpark of ~35k bp. We can filter them a bit:
jq '.reports[] | select(.completeness == "COMPLETE") | .length' adenovirus.json
24633
24630
24659
Three of the genomes are considered complete. How about we filter based on length?
jq '.reports[] | select(.length >= 30000) | "\(.completeness) \(.length)"' adenovirus.json | wc -l
This gives us 82 genomes - interestingly enough, although longer than the "complete" genomes, these are considered incomplete.
It turns out there are lots of adenoviruses. I'm interested in the mastadenovirus, which are the ones that infect humans. There are bunch of different types, A-G. We can search with a different term, "HAdV", whihc Human Adenovirus**.
I didn't find anything - datasets gave me an error:
Error: The taxonomy name 'hadv' is not a virus taxon. Try using one of the suggested taxids:
Human mastadenovirus B (species, taxid: 108098, HAdV-B)
Human mastadenovirus C (species, taxid: 129951, HAdV-C)
Human mastadenovirus D (species, taxid: 130310, HAdV-D)
Human adenovirus 81 (serotype, taxid: 1972755, HAdV 81)
Human adenovirus 56 (no-rank, taxid: 880565, HAdV-D56)
Human mastadenovirus F (species, taxid: 130309, HAdV-F)
Human adenovirus 61 (no-rank, taxid: 1069441, HAdV 61)
Human adenovirus 65 (serotype, taxid: 1094363, HAdV 65)
Human mastadenovirus E (species, taxid: 130308, HAdV-E)
Human mastadenovirus A (species, taxid: 129875, HAdV-A)
I tried "human adenovirus". I got a bunch of hits, 20 of which were complete!
jq '.reports[] | select(.completeness == "COMPLETE") | .length' hadv.json | wc -l #=> 20
We can grab the accession:
jq '.reports[] | select(.completeness == "COMPLETE") | .accession' hadv.json
"PQ164815.1"
"OL450401.1"
"MW306919.1"
"MT113944.1"
"MT113943.1"
"MT113942.1"
"MK570619.1"
"MK570618.1"
"MK357715.1"
"MK357714.1"
"MK241690.1"
"LC215437.1"
"LC215436.1"
"LC215435.1"
"LC215434.1"
"LC215433.1"
"LC215429.1"
"KY002685.1"
"KY002684.1"
"KY002683.1
I ended up writing some python to map these to the groups (A-G). It turns out:
KY => B MT => B LC => D MW => E
Now, let's get the genomes. We can visit the direct link by going to https://www.ncbi.nlm.nih.gov/nuccore/{entry}, but we can grab them all using NCBI Entrez.
We can ask for the nucleotides in fasta format!
efetch -db nucleotide -id PQ164815.1 -format fasta
# or, all at once:
for acc in $(jq -r '.reports[] | select(.completeness == "COMPLETE") | .accession' hadv.json)
do
efetch -db nucleotide -id ${acc} -format fasta > ${acc}.fasta
done
Weirdly, the fasta files are incorrectly formatted? The first line is
>OL450401.1 Human adenovirus sp. isolate MKC_4, complete genom
Should be a space after the >. I fixed them up:
#!/bin/bash
# Loop over all .fasta files in the current directory
for file in *.fasta; do
# Use sed to add a space after '>' if there isn't one
sed -i 's/^>\([^ ]\)/> \1/' "$file"
done
Then, put two into a single file, to run clustalo, found here.
Before we do, let's look at what we are dealing with:
>OL450401.1 Human adenovirus sp. isolate MKC_4, complete genome
>PQ164815.1 Human adenovirus sp. isolate HAdV-JUH23/KSA/2024, complete genome
According to NCBI, the first is Adenovirus D. The second doesn't say.
Now, run clustalo:
# 2 sequences, about 1m on my machine
clustalo -i all.fasta -o aligned.aln --outfmt=clustal
Doing them all will take a long time. I did 2 here. I also did all the ones starting with LC*, like this:
# 6 sequences, about 1m on my machine
cat LC2154* > LC.fasta
clustalo -i LC.fasta -o LC.aln --outfmt=clusta
I used --outfmt=clustal since I found the BioPython library, which I use for the consensus sequence, doesn't work with the default output from clustalo.
Identifying Regions of Interest
Using the MSA from the two genomes above, here's some useful things we might do.
Generate a Consensus Sequence
We can generate a consensus sequencing using BioPython:
from Bio import AlignIO
from Bio.Align import AlignInfo
alignment = AlignIO.read("../aligned.aln", "clustal")
summary_align = AlignInfo.SummaryInfo(alignment)
consensus = summary_align.dumb_consensus()
print(consensus) # around 38k nt
Not much use (in isolation, at least).
Shannon Entropy
We can compare N genomes from our MSA and see how conserved each position is:
import numpy as np
from collections import Counter
from Bio import AlignIO
def shannon_entropy(column):
counts = Counter(column)
freq = np.array(list(counts.values())) / sum(counts.values())
return -np.sum(freq * np.log2(freq))
alignment = AlignIO.read("../aligned.aln", "clustal")
entropies = [shannon_entropy(column) for column in zip(*alignment)]
breakpoint()
import matplotlib.pyplot as plt
plt.plot(entropies)
plt.xlabel("Genome Pos")
plt.ylabel("Shannon entropy")
plt.title("Genomic Variability in Adenovirus")
plt.show()
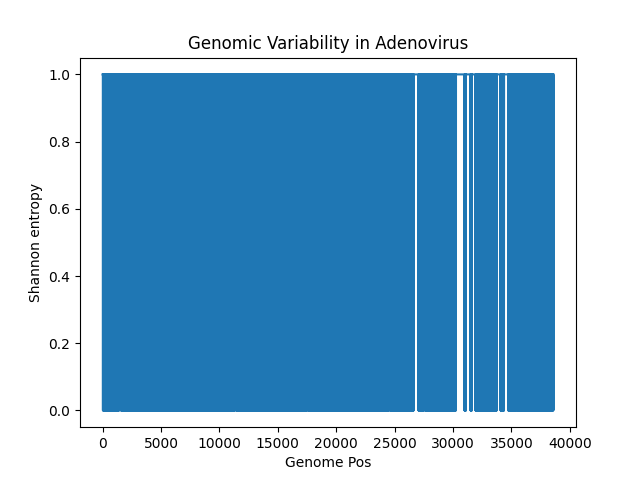
Almost the entire thing is 1 - there is hardly any conserved regions. I think that's because they are two different strains of Adenovirus, which has a lot of variability.
I did the same thing for the LC* samples mentioned earlier - I checked them all on NCBI, those are all Adenovirus D.
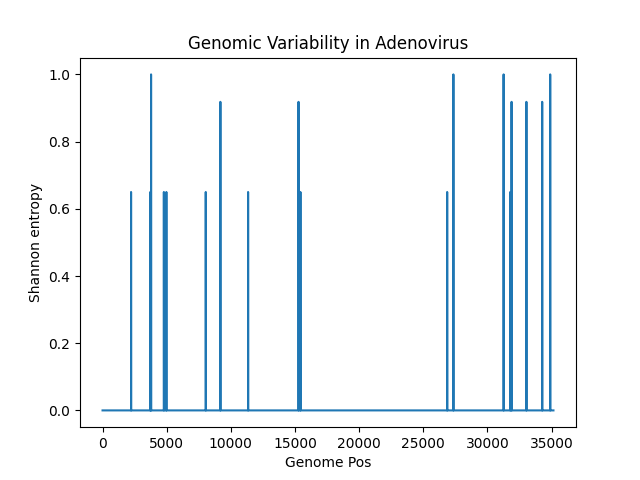
Adenovirus F
I am paricularly interested in Adenovirus F. The JSON file we got with datasets doesn't tell us the serotype, though. We can get some more information via the NBCI data, though, with a nifty python script:
accessions = [
"PQ605050.1",
# ...
"KY002684.1",
"KY002683.1",
]
from Bio import Entrez
from Bio import SeqIO
import json
import time
Entrez.email = "lachlan@lachlan-miller.me"
def fetch_serotype(accession):
# don't hammer the server
time.sleep(0.2)
try:
# Fetch the GenBank record from NCBI
with Entrez.efetch(db="nucleotide", id=accession, rettype="gb", retmode="text") as handle:
record = SeqIO.read(handle, "genbank")
# Search for the serotype in the features
for feature in record.features:
if feature.type == 'source':
with open(f"data/{accession}.txt", "w") as f:
if feature.qualifiers:
out = json.dumps(feature.qualifiers, indent=4)
print(accession, out)
f.write(out)
except Exception as e:
print(f"Error fetching {accession}: {e}")
for idx, accession in enumerate(accessions):
print(f"{idx} / {len(accessions)}")
fetch_serotype(accession)
We get a bunch of information, eg:
{
"organism": [
"Human adenovirus sp."
],
"mol_type": [
"genomic DNA"
],
"strain": [
"HAdV-F41/IAL-AD10449/2006/BRA"
],
"isolate": [
"IAL-AD10449/06"
],
"isolation_source": [
"feces"
],
"host": [
"Homo sapiens"
],
"db_xref": [
"taxon:1907210"
],
"geo_loc_name": [
"Brazil"
],
"collection_date": [
"2006"
],
"note": [
"group: F; genotype: 41"
]
}
Where the note and strain tell us this is Adenovirus F. Not every dataset is as clear:
{
"organism": [
"Human adenovirus sp."
],
"mol_type": [
"genomic DNA"
],
"strain": [
"HAdV/S154/Huzhou/2021/CHN"
],
"isolate": [
"S2021154"
],
"isolation_source": [
"stool sample"
],
"host": [
"Homo sapiens"
],
"db_xref": [
"taxon:1907210"
],
"geo_loc_name": [
"China"
],
"collection_date": [
"17-Jun-2021"
]
}
Looking closer on NCBI:
LOCUS OL897264 761 bp DNA linear VRL 15-NOV-2022
It's only 761bp - can't say much with such a tiny snippet of DNA.
I grabbed the nucleotides for each one with genotype: F41:
grep -ir "genotype: F41" | sed 's/\.txt:.*//' | while read id; do
echo "fetching $id..."; efetch -db nucleotide -id "$id" -format fasta > "../adenovirus_f_snippets/$id.fasta"
After some more wrangling, I found a bunch of sequences around 474 nucleotides long, each one labeled
Human adenovirus sp. isolate TUN/20778/HAdV-41/2014 hexon gene, partial cds
Looking one of the sequences up: https://www.ncbi.nlm.nih.gov/nuccore/OP078715.1/
There's plenty of information about the Adenovirus hexon gene.
I did a BLAST - this gene looks to be a compelling diagnostic marker for Adenovirus F (type 41).